
周末文摘 | 我国创新药审评审批的改革与发展——以抗肿瘤药物为例
发布时间:2024-09-26 12:31分类: 无 浏览:100评论:0


引用本文

周思源,杨志敏,宋媛媛,黄果*.我国创新药审评审批的改革与发展——以抗肿瘤药物为例[J].中国食品药品监管.2024.06(245):4-15.
我国创新药审评审批的改革与发展
——以抗肿瘤药物为例
Reform and Development of Innovative Drug Review and Approval in China: Taking Anti-Tumor Drugs as an Example
周思源
国家药品监督管理局药品审评中心
ZHOU Si-yuan
Center for Drug Evaluation,National Medical Products Administration
杨志敏
国家药品监督管理局药品审评中心
YANG Zhi-min
Center for Drug Evaluation,National Medical Products Administration
宋媛媛
国家药品监督管理局药品审评中心
SONG Yuan-yuan
Center for Drug Evaluation,National Medical Products Administration
黄果*
国家药品监督管理局
HUANG Guo*
National Medical Products Administration
摘 要
Abstract
近年来,我国药品监管部门积极采取多项措施,旨在加快创新药的上市速度,以满足广大患者迫切的医疗需求。通过实施附条件批准程序、突破性治疗药物程序、优先审评审批程序等多种加快药物审评审批路径,有效推动了药物研发进程。同时,药品监管科学的不断进步也为此提供了坚实的技术支撑。本文以10 余年来我国抗肿瘤药物审评技术的演进与创新为例,阐述了创新药审评评价体系的建立与完善,以期为抗肿瘤药物的审评和研发工作提供指导。未来,药品监管部门将继续致力于推动监管创新,并特别关注罕见肿瘤、儿童肿瘤等领域的创新药研发,坚持完善审评审批体系和提高审评效率,保障广大患者能够获得更多优质、安全、有效的治疗药物。
In recent years, the drug regulatory authorities of China have been proactive in implementing various measures aimed at expediting the marketing approval of domestically developed innovative drugs and imported drugs already marketed overseas. This is done to address the urgent medical needs of the vast majority of patients. Through the implementation of conditional approval procedures, breakthrough treatment drug identification, priority review and approval and other accelerated drug review and approval paths, it provides a strong guarantee for the accelerated innovation of domestic anti-tumor drugs, and effectively promotes the research and development process of anti-tumor drugs. Concurrently, the ongoing advancement of drug regulatory science has offered solid technical backing for drug research and development. The gradual establishment and refinement of China's anti-tumor drug evaluation system have provided more scientific and standardized guidance for drug evaluation and development. This paper presents a retrospective review of the evolution and innovation of evaluation standards in China over the past decade. The drug regulatory authorities will persist in promoting the regulation of anti-tumor drug innovation, paying particular attention to rare tumors and pediatric tumors in the realm of drug research and development in future. This will be achieved by refining the review, examination, and approval system and enhancing the efficiency of evaluation reviews, striving to ensure that tumor patients can access more high-quality, safe, and effective treatment options.
关键词
Key words
药品监管;抗肿瘤药;审评审批;技术标准
drug regulatory; anti-tumor drugs; review and approval; technical standards
近年来,我国药物研发取得了突飞猛进的发展,并始终保持着旺盛的创新活力,众多领域实现从无到有的重大突破,显著缩小了与世界顶尖治疗产品技术水平的差距,极大地满足了患者的治疗需求。同时,药物研发模式也经历了深刻的变革:国产药由最初的仿制药为主导,逐步向创新药领域拓展;进口药则由原先的滞后引进,逐步向全球同步研发过渡。此外,随着研发实力的提升,国内制药企业开始尝试License-out 授权(海外企业进行后期研发和销售)的研发模式,使我国的创新药逐步走向世界,为全球更多患者提供治疗选择。与此相伴,我国药品审评能力显著提升。药品监管部门持续深化药品审评审批制度改革,着力构建一套科学且完备的药品审评质量管理规范(good reviewpractice,GRP),以整合各方资源,优化审评流程,确保审评工作及其管理的质量与标准相统一。同时,通过提高审评效率和透明度,进一步明晰了审评流程,为公众提供更加清晰、透明的药品审评信息,为药品加快上市提供有力保障。
本文以抗肿瘤药物发展为例,总结了我国药品审评审批制度改革在推动药物研发上市中取得的进步和成果。可以看到,我国抗肿瘤药物研发实现了从细胞毒性药物到小分子靶向药物,再到免疫治疗药物,以及在近年来备受瞩目的细胞治疗药物等领域的重大突破,在这个过程中,我国抗肿瘤药物审评评价体系建设、注册申请路径创新、临床审评技术演进为抗肿瘤药物的研发创新提供了坚实的技术指导和有力的制度保障。
1 我国抗肿瘤药物审评评价体系的建设
2018 年以前,国产药物以仿制药为主。2018年开始,创新药批准数量呈逐年递增趋势,特别是国产创新药数量较之前明显增多,其中2021 年国产新药的上市数量更是首次超过了进口药。尤其是随着药物研发技术、精准医学等基础研究不断深入,我国抗肿瘤药物领域迅速发展,这不仅对药物研发企业提出了更高的要求,也对药品监管部门提出了新的挑战。
为应对这些挑战,一方面,我国药品监管部门积极对接国际药品监管标准。自2017 年我国药品监管部门正式成为国际人用药品注册技术协调会(The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,ICH)的成员国以来,致力于将ICH 的指导原则在国内转化实施。目前,国家药监局已经基本完成了ICH 当下全部指导原则的落地实施工作,这一里程碑式的成就标志着我国在药品注册申请、审评审批、质量控制等核心领域已经与国际接轨。同时,我国还积极参与ICH 的各项活动,为推动全球药品监管体系的完善贡献了中国智慧和中国方案,展现大国担当,显著提升了我国在全球药品监管领域的地位和影响力。
另一方面,为解决药物研发企业在药品研发过程中的技术性难题,国家药品监督管理局药品审评中心(以下简称药审中心)制定并发布了一系列指导原则,进一步完善抗肿瘤药物的审评评价体系。截至2024 年3 月,药审中心已累计发布技术指导原则503 项,其中肿瘤适应症相关指导原则61 项。这些指导原则的制定和发布,为药物研发企业提供了明确的技术指导和规范,在指引抗肿瘤创新药物的研发方向、加速境外已上市抗肿瘤药物的引进流程以及推动儿童抗肿瘤药物的研发和上市等多个关键环节,起到了至关重要的作用。
2 我国抗肿瘤药物注册申请路径的创新
加快药品审评审批的速度,是药品监管部门对公众关于提高审批效率需求的及时回应,也是满足临床迫切需求的关键措施。2020 年版《药品注册管理办法》新增了附条件批准程序、突破性治疗药物程序、优先审评审批程序及特别审批程序,形成了我国药品加快上市注册的“四条路径”。这项改革促进了以临床价值为导向的药品研发,特别是在抗肿瘤药物这种创新性更强、研发速度更快的领域,为研发机构提供了在市场竞争中取得先机的可能,为推动行业创新注入了新的活力。尤为重要的是,针对肿瘤这类严重威胁公众生命健康的疾病,加速新药的研发与上市为患者带来的是生存的希望。
在“四条路径”中,附条件批准是最具开创性的,也是鼓励药物创新研发的最有力措施之一。这一概念的引入,可追溯至2016 年2 月发布的《总局关于解决药品注册申请积压实行优先审评审批的意见》(已废止),该文件首次提出了“有条件批准上市”的情形,是首次在我国引入药品非完全批准上市的概念。随后,在2017 年10 月发布的《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》[1] 中,采用了“附带条件批准”的表述。新修订《药品管理法》[2] 则在法律层面正式确立了附条件批准的制度框架。基于药品研发与审评的实际操作经验,2020 年版《药品注册管理办法》[3] 明确规定了可以申请附条件批准的多种情形。此外,为了进一步明确工作机制和技术标准,还相继发布了《药品附条件批准上市申请审评审批工作程序(试行)》[4] 和《药品附条件批准上市技术指导原则(试行)》[5]。据统计,截至2024 年3 月,我国获得附条件批准的抗肿瘤药物已有70 种(涵盖82个抗肿瘤适应症)。
在加快审评审批的同时,后续的监管措施也在不断加强。例如,针对附条件批准后如何有效督促申请人及时完成确证研究的问题,各国监管部门都在积极探索。2022 年12 月,美国通过了《2022 年食品和药品综合改革法案》(The Food and Drug Omnibus Reform Act of 2022), 该法案赋予了美国食品药品监督管理局(Food andDrug Administration,FDA)撤回加速批准的权力。在我国,药品注册证书的常规批准有效期设定为5 年[3],因此,对于附条件批准的药品注册证书,其有效期亦不得超过5 年。但是,由于补充应用验证性试验数据的审评时限为200 天,因此,申请人原则上需在附条件批准后的4 年内提交相关数据。2023 年8 月25 日,国家药监局公开征求了《药品附条件批准上市申请审评审批工作程序(试行)(修订稿征求意见稿)》[6] 的意见,明确指出,上市后需继续完成的研究工作,其完成时限原则上不超过4 年;对于附条件批准上市的品种/ 适应症,只有完成所附条件研究并将批准转为完全批准后,方可启动再注册工作。此举旨在确保药品的安全性和有效性,平衡抗肿瘤药物的风险和收益。
3 我国抗肿瘤药物临床审评技术的演进
强大的监管助力高质量发展,药品监管科学的进步为药物研发提供有力支撑。我国抗肿瘤药物审评评价体系的逐步建立与完善,正是药品监管科学与药物研发相互促进的成果。
3.1 单臂试验为无药可治的患者尽早提供治疗机会
20 世纪20 年代初期,随机化概念被引入科学研究领域。自20 世纪60 年代起,为确保药物的有效性评价具有科学性和可靠性,随机对照试验(randomized controlled trial,RCT)被广泛应用,并成为公认的评估药物有效性的金标准。
在细胞毒类抗肿瘤药物治疗的背景下,抗肿瘤药物的研发模式主要遵循3 个阶段的临床试验流程:①Ⅰ期临床试验聚焦于末线肿瘤患者,通过逐步增加药物剂量的方式,探索试验药物的最大耐受剂量(maximal tolerable dose,MTD)。②Ⅱ期临床试验在已确定的安全剂量范围内,选择一个尽可能高的剂量,针对潜在有应答的肿瘤患者人群进行扩展研究,从而完成概念验证。③Ⅲ期临床试验在目标患者群体中开展一项RCT 研究,为药物的上市提供有力支持。这种严谨的研发流程确保了抗肿瘤药物的安全性和有效性,为患者提供了更为可靠的治疗选择。
尽管RCT 在药物研发中具有重要地位,但其对样本量和研究时间有较高要求,使临床急需的抗肿瘤药物上市需要经历较长时间。因此,在某些特殊情况下,药品监管部门必须考虑采用没有对照组的单臂试验(single arm trial,SAT)作为审评依据。例如,我国首个基于单臂研究申报上市的药品西达本胺(英文名称:Chidamide,英文别名:CS055)于2014 年获得批准,用于治疗复发难治的外周T 细胞淋巴瘤(peripheral T cell lymphomas,PTCL)[7]。
在西达本胺的研发过程中,国内针对复发难治的PTCL 尚未有疗效确切的药物问世。西达本胺通过Ⅰ期临床试验初步评估了安全性和耐受性,并为其后续给药剂量提供了研究依据[8]。在此基础上,申请人、临床专家与药审中心共同确定了采用多中心、单臂、非随机、开放的研究设计方案, 以客观缓解率(objective response rate,ORR)作为主要研究终点,设定了ORR 不低于27%,95% 置信区间(confidence interval,CI)下限不低于15% 的目标,并要求由独立评审委员会(Independent Review Committee,IRC)对其有效性进行评价。研究结果显示,79例复发难治的PTCL 受试者在接受西达本胺治疗后, 经IRC 评估ORR 为27.8%(95%CI :18.3%~39.0%),达到了试验预设[9]。西达本胺上市后,一项针对复发难治的PTCL 的真实世界研究结果显示,西达本胺的ORR 为39.06%,体现了较好的一致性[10]。继西达本胺之后,多个国内外药物通过单臂研究得以附条件批准上市。
为了规范药物研发单位合理地应用单臂临床试验设计,2020 年药审中心制定发布了《单臂试验支持上市的抗肿瘤药进入关键试验前临床方面沟通交流技术指导原则》[11] 和《单臂试验支持上市的抗肿瘤药上市许可申请前临床方面沟通交流技术指导原则》[12] 两项指导原则。2023 年,又发布了《单臂临床试验用于支持抗肿瘤药上市申请的适用性技术指导原则》[13],系统阐述了当前对单臂临床试验用于支持抗肿瘤药物上市申请的适用性的科学认识。
单臂临床试验的应用更加科学审慎。单臂临床试验作为关键性研究的前提,必须是药物的作用机制清晰明了。对于罕见肿瘤这类难以实施对照研究的疾病,只有在药物疗效非常突出的情况下,单臂临床试验才可能是合适的临床试验设计。否则,应更多采用对照研究,借助中间临床终点或替代终点[ 例如ORR、完全缓解率(complete response rate,CRR)等] 来支持药物的附条件批准上市,之后再通过同一研究的长期随访或重新开展一项RCT 的无进展生存期(progression-free survival,PFS)或总生存期(overall survival,OS)等结果,进一步确证药物的有效性和安全性,以支持其完全批准。
3.2 生物标志物为患者带来精准治疗
早期,肿瘤治疗主要依赖于细胞毒性药物,但缺乏特异性。然而,随着靶向治疗和免疫治疗的发展,肿瘤治疗逐渐变得更加精准。这种精准不仅体现在药物更准确地针对肿瘤细胞,还体现在更准确地定位获益人群。
经过精确识别目标治疗人群后,药物的有效性通常会得到显著提升。对于药物研发企业而言,精确筛选潜在受益人群,可以进一步凸显药物的有效性,进而大幅提高药物临床研发的成功率和药品上市的速度。生物标志物作为一种重要的工具,有助于特定人群的富集。鉴于此,2021 年药审中心发布了《生物标志物在抗肿瘤药物临床研发中应用的技术指导原则》[14],详细阐述了生物标志物的定义和分类,并重点强调了生物标志物在抗肿瘤药物有效性和安全性研究中的应用。
值得注意的是,与采用阳性与阴性二分法定性评估的生物标志物不同(即阳性患者接受治疗后效果良好,而阴性患者则无明显疗效),部分药物疗效与生物标志物的表达水平紧密相关。例如,细胞程序性死亡配体1(programmed cell death-ligand 1,PD-L1)的表达水平对PD-1/PD-L1抑制剂的治疗效果有明确影响。为评估PD-L1 表达水平,学界制定了例如肿瘤细胞阳性比例分数(tumor proportion score, TPS)、联合阳性分数(combined positive score, CPS)等评价标准。通过精确测定PD-L1 表达水平,可以为免疫检查点抑制剂的应用提供重要指导。
以帕博利珠单抗为例,研究结果显示,对于PD-L1 TPS ≥ 50% 的一线非小细胞肺癌(nonsmallcell lung cancer,NSCLC) 患者, 使用帕博利珠单抗治疗,经过5 年的随访,中位OS达到了26.3 个月,5 年生存率高达31.9%,该疗效显著优于现有的化疗方案[15]。因此,如果选择TPS ≥ 50% 的NSCLC 作为目标适应症,疗效可能更突出,研发成功率更高,甚至可能加速药物的上市时间。
随着分子诊断学的不断进步,能精准识别驱动肿瘤疾病生物学改变的核心基因变异成为了可能,进而改变了临床治疗的整体策略。肿瘤遗传学的研究亦揭示出其复杂性,致癌变异通常缺乏特异性,致癌基因可在多种癌症类型中被共享。第二代测序技术在临床肿瘤学中的普及,为检测这些跨肿瘤类型的分子生物标志物提供了有力工具,从而催生了“泛肿瘤”这一新兴概念。“泛肿瘤”作为一种新颖的疾病认知框架,从肿瘤起源与发病机制的角度,将源自不同组织类型的肿瘤视作一种统一疾病[16],并致力于探索普适性的治疗手段。
2017 年5 月,FDA 正式批准全球首款针对非特定瘤种的“广谱抗肿瘤药物”——帕博利珠单抗,用于治疗具有微卫星高度不稳定(microsatellite instability-high,MSI-H)或错配修复基因缺陷(deficient mismatch repair,dMMR)特征的实体瘤患者[17]。这一批准不仅拓宽了肿瘤治疗的领域,也预示着肿瘤治疗策略从传统的基于人体起源组织,逐渐转变为依据肿瘤遗传特征的个性化治疗。自此之后,国内外医药企业纷纷跟进,研发并上市了多款不限瘤种的抗肿瘤药物。截至2024 年2 月,我国已批准了7 款不限瘤种的抗肿瘤药物(表1),这些药物的陆续上市不仅体现了临床实践、基础研究与药品监管衔接的不断进步,更彰显了我国肿瘤治疗领域从组织起源向遗传特征的重要转变。

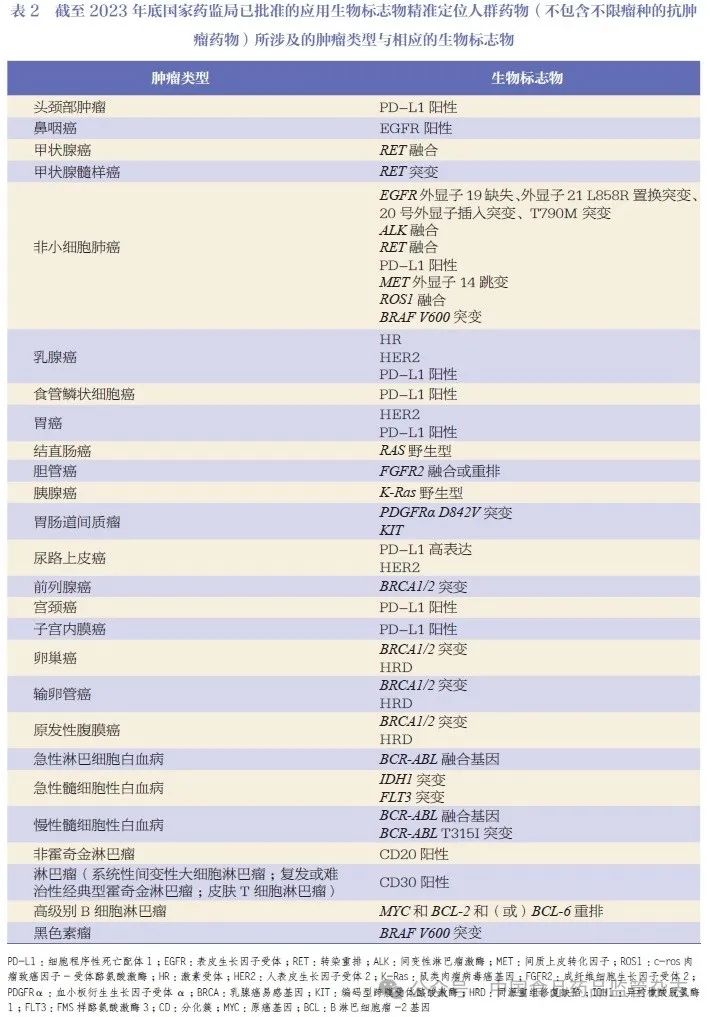
此外,截至2023 年底,我国已有新药73 个和改良型新药的109 项上市申请,使用生物标志物来精准定位目标治疗人群,涉及多个适应症领域。本文对所针对的肿瘤类型及其相应的生物标志物进行了列举,见表2。
3.3 以患者为中心的人群多样化
相对于富集人群,在临床研发过程中,须充分考虑到患者群体的多样性,包括处于不同疾病阶段的患者以及特殊人群的治疗需求。这种兼顾不同需求的策略,彰显了药品审评审批工作对治疗全面性和普适性的追求。
在抗肿瘤药物的适应症开发和申报过程中,通常遵循从末线人群逐步向前线人群推进的规律。具体而言,研究人员一般以末线患者为基础,而前线患者群体的适应症开发相对滞后。然而,这种研发策略导致前线患者或初治患者无法及时获得新药物和新疗法,这与肿瘤治疗中强调的“早发现、早治疗”原则相悖。因此,虽然早期探索研究需要在末线患者中开展,但当在这部分患者中观察到试验药物具备疗效时,即可开始着手在前线或初治适应症中的开发。以免疫治疗为例,针对尚未出现免疫功能严重衰退的早期患者,其治疗效果可能更加显著,此时若及时开发应用于早期肿瘤阶段的相关适应症,对于患者而言,无疑具有更高的临床价值。
各类肿瘤患者年龄分布特征各异,身体器官功能状态亦可能因年龄、病情等差异受影响。随着抗肿瘤药物研发与上市的快速推进,部分研发企业在临床试验中可能因经验不足而忽略药物在特定人群中的研究。鉴于此,药审中心在日常审评及沟通工作中,会强调并提醒研发企业在药物研发的起始阶段和关键节点,及时完成相关研究。
3.4 对照组的升级确保患者获得更优治疗
在我国,化疗仍是多数肿瘤的标准治疗,在抗肿瘤新药的临床试验中也通常采用化疗作为对照组。随着新药的不断研发和上市,我国临床实践中的标准治疗也在逐步演进。如果关键研究仍然以原有的标准治疗(例如化疗)作为对照,将难以充分展现新药的临床价值。针对这种临床实践背景和研发环境的变化,药审中心发布了《以临床价值为导向的抗肿瘤药物临床研发指导原则》[18],明确指出新药研发的核心目标应为患者提供更优质的治疗选择,包括更高的有效性、安全性和便利性。同时,应重视患者治疗需求的动态变化,对照药物的选择应能反映并代表临床实践中目标患者的最佳治疗方案。因此,在药物研发过程中,须审慎考虑对照药物的选择,以确保新药能够真正满足患者的治疗需求,实现其临床价值。
以晚期NSCLC 一线治疗为例,含铂化疗一直是其标准治疗方案。在我国,多个PD-1 单抗的Ⅲ期关键研究均选择了含铂化疗作为对照组。随着国产PD-1 单抗陆续在国内上市,免疫治疗在NSCLC 领域的应用日益广泛,并逐步取代了含铂化疗,成为晚期NSCLC 一线治疗的“新标准”。因此,在免疫治疗背景下,药审中心已要求将PD-1 联合化疗作为晚期NSCLC 一线治疗对照研究的新对照组。在同类靶点、同机制药品已上市的背景下,为确保新药临床价值的体现,临床试验中可能需要将已上市的同类产品作为对照,或将经过已上市同类药物治疗后出现的难治或复发患者群体作为目标适应症人群,以体现新药的临床价值。
临床价值不仅体现于关键研究中对照药的遴选,更应贯穿于药物研发的每个环节。在新药研发的众多领域中,双特异性抗体(bispecific antibody,BsAb)备受瞩目。据统计数据显示,全球临床试验数量呈现出持续上升的趋势,截至2019 年3 月,全球商业化临床研究阶段的BsAb药物数量已超过85 个,其中针对肿瘤适应症的BsAb 药物占比超过85%[19]。
恶性肿瘤的发生、发展涉及多种病理组织学和分子生物学机制,这使得仅针对单一靶点的单克隆抗体(单抗)通常难以达到理想的治疗效果。BsAb 的开发旨在提供相较于单抗更大的临床优势,并可能在与其他治疗或复方制剂的联合应用中表现出优势,但BsAb 在结构和生产工艺上相较于单抗都更为复杂。为引导药物研发机构进行科学、合理的BsAb 设计,避免盲目开发,药审中心制定了《双特异性抗体抗肿瘤药物临床研发技术指导原则》[20]。该指导原则强调,抗体设计应以临床需求为导向,并要求如果在相同适应症的当前最优标准治疗中,若已包含BsAb 中的任一相同靶点单抗单药或联合用药时,则在随机对照研究中,应选择含该单抗的标准治疗方案(单药或联合用药)作为对照药;若任一靶点具备单抗成药性,则需审慎评估开发BsAb 的必要性和合理性。必要时,还需开展与单抗的对照研究。
3.5 终点指标更加体现临床获益
在评估抗肿瘤药物疗效方面,OS 一直被视为金标准。随着众多具备不同作用机制的抗肿瘤药物陆续获得批准并投入市场,肿瘤患者生存期不断延长,使某些肿瘤逐渐呈现出慢性疾病的特点。生存期的延长,意味着在临床试验中,若以OS 作为主要疗效评价指标,所需的观察与随访时间将显著增长,则可能导致药物上市时间相应推迟。同时,后续治疗选择变得更加丰富,病情变化也日趋复杂,这些因素都为OS 评估增加了干扰。因此,为加速肿瘤药物的研发进程,研发过程中采用经过合理验证的替代终点显得尤为重要。
2020 年版《药品注册管理办法》[3] 及配套发布的《药品附条件批准上市技术指导原则(试行)》[5]指出,替代终点的选择需基于其对临床获益的预测能力。具体来说,如果替代终点是已知能够合理预测临床获益的指标,可被用于常规批准;如果替代终点是有可能预测临床获益的指标,可被用于附条件批准。然而,通过对采用替代终点获得批准的药物临床试验后续结果进行回顾性研究,可以发现替代终点的潜在获益并不总是能够转化为患者的实际生存获益。因此,选择何种终点作为替代终点,以及在药物研发过程中如何恰当地运用替代终点,显得尤为关键。此外,各种替代终点具有其独特的功能特性,有必要结合疾病的特性、药物的作用机制、不同临床试验阶段以及疾病的分期来科学合理地应用替代终点。
以早期的急性淋巴细胞白血病(acute lymphoblastic leukemia,ALL)/ 急性髓细胞性白血病(acute myeloid leukemia,AML)为例,其治疗目标是获得深度的缓解,以赢得造血干细胞移植的机会,因此在这类案例中,CRR 可能被视作合适的替代终点。对于血液肿瘤而言,相较于实体瘤,其病理取材过程相对简便,为其以微小残留病(minimal residual disease,MRD) 这一替代终点的开发提供了潜在优势。为此,药审中心先后发布了《急性淋巴细胞白血病药物临床试验中检测微小残留病的技术指导原则》[21]、《慢性髓细胞白血病药物临床试验中检测微小残留病的技术指导原则》[22]、《多发性骨髓瘤药物临床试验中应用微小残留病的技术指导原则》[23] 等多项技术指导原则。近期,FDA 亦在肿瘤药物专家委员会(Oncologic Drugs Advisory Committee)会议上讨论了未来利用MRD 支持多发性骨髓瘤领域加速批准的可能性[24]。
一些疾病进展较快,PFS 的显著延长本身就是临床获益,因此,在这些情况下,将PFS 作为主要研究终点也是合理的。以首个国产CDK4 和CDK6 抑制剂达尔西利为例,在一项针对既往内分泌治疗无效的激素受体阳性(HR+)晚期乳腺癌患者的Ⅲ期随机对照研究中,达尔西利联合氟维司群相较于单药氟维司群,显著延长了患者的PFS(15.7 个月vs 7.2 个月,风险比为0.42),同时OS 也看到了获益趋势[25]。基于该研究结果,国家药监局于2021 年批准了达尔西利的上市,为患者提供了新的治疗选择。
处于不同疾病阶段的肿瘤开展临床试验时,对于试验终点的考量可能也存在差异。为此,药品监管部门先后发布了《晚期非小细胞肺癌临床试验终点技术指导原则》[26]、《晚期肝细胞癌临床试验终点技术指导原则》[27] 等指导原则,阐述了在不同的晚期瘤种中不同疾病阶段替代终点运用的考虑。
不同药物作用机制也是在选择终点时需要考虑的重要因素。在既往的化疗及靶向治疗药物的临床试验中,PFS 与OS 的相关性较强,因此常被用作替代终点。以抗体偶联药物(antibody-drug conjugate,ADC)为例,其类似于化疗药物,在有效性评估中,可将PFS 作为主要终点,而OS作为次要终点。与之不同的是,在免疫治疗药物的临床试验中,鉴于免疫检查点抑制剂的起效具有延迟性,PFS 将难以准确预测OS 的获益。因此,在免疫治疗药物的临床试验中,以OS 作为主要终点可能更为适宜。近年来,我国临床专家还提出了一种对传统PFS 的改良研究设计,该研究设计在统计上排除了3 个月内的早期疾病进展事件,旨在最大化PFS 与OS 之间的相关性[28-29]。这为我们提供了新的视角,并为开发新的替代终点提供了思路。
新药研发的核心目标是患者利益最大化。对于肿瘤患者而言,延长其生存期仍是当前最关键的目标。然而,肿瘤疾病本身及其治疗手段均可能给患者带来沉重的疾病负担,进而严重影响其生活质量。因此,在药物研发过程中,对患者生存质量的提升和生活体验的改善受到了越来越多的关注。根据《以临床价值为导向的抗肿瘤药物临床研发指导原则》[18],鼓励申请者运用生活质量评估、症状评估等患者报告结果(patient reported outcomes,PRO)工具,深入了解药物治疗在缓解患者症状和提高生活质量方面的效果;倡导在肿瘤患者群体中收集PRO 数据,以获取无法通过其他传统评价指标(例如OS、PFS)所揭示的重要临床信息。这些数据能够更全面地反映抗肿瘤药物治疗对患者的影响,以及治疗对患者生活质量的提升作用。
4 展望
通过加速审评程序获批上市的新药,正在重塑肿瘤治疗格局,为我国肿瘤患者带来更为丰富多样的治疗选择。从传统的化疗、放疗、手术模式,到现今的单抗、靶向治疗、BsAb、ADC 类药物以及嵌合抗原受体T 细胞免疫治疗(chimeric antigen receptor T cell immuno-therapy,CAR-T)等,这些治疗手段更为精准和多元,显著延长了患者的生存时间。
尽管取得了成效,但我国的药物研发创新仍面临着巨大的挑战。以抗肿瘤药物为例,随着国家经济实力的增强和公众生活质量的提升,我国居民疾病谱系发生了显著变化,自2006 年起,恶性肿瘤已成为我国首要健康威胁[30]。同时,我国已经进入中度老龄化社会,随着老龄化进程加速推进,癌症负担可能会进一步加剧[31]。据全球统计数据显示,2018 年有1810 万人新诊断为癌症,其中960 万人因癌症去世。在这种背景下,我国以新增病例430 万例(占全球总数的24%)和死亡病例290 万例(占全球总数的30%)的数据,成为全球癌症负担最重的国家之一[32],加速研发上市抗肿瘤药物成为满足患者临床需求的重大挑战。
未来,我国药品监管部门将继续积极推动监管改革创新,以适应包括抗肿瘤药物在内的不断发展的临床治疗需求和药物研发需求。首先,持续完善技术评价体系以确保药物安全性和有效性。药品监管部门将持续关注国际药品监管的最新动态和技术进展,引进和借鉴国际先进的评价方法与技术手段,不断完善药物审批流程。加强与国内外药品研发机构的沟通与合作,共同推动药物技术创新和质量提升。其次,持续关注罕见肿瘤、儿童肿瘤等“冷门”领域。这些领域的疾病发生率相对较低,但患者通常面临更为严峻的治疗挑战。药品监管部门将持续引导和鼓励相关企业深入这些领域的药物研发,通过政策支持和监管创新,提高研发效率,为患者提供更多有效的治疗选择。此外,持续加强与临床医生的沟通与合作。深入了解临床需求和实际疗效,为药物审评审批提供更为科学、合理的依据,并积极推动药物的研发与临床应用相结合,形成产学研用一体化的良性循环。着力推进细胞治疗等前沿治疗方法在肿瘤领域的应用,促进我国药物领域的全面发展。
总之,我国药品监管部门在未来将继续深化药品审评审批制度改革,持续推动监管创新,为患者获得更多优质、安全、有效的治疗药物保驾护航。
第一作者简介
周思源,国家药品监督管理局药品审评中心主任、党委书记。专业方向:药品审评
通讯作者简介
黄果,国家药品监督管理局党组成员、副局长,第十二届药典委员会副主任委员。专业方向:药品监管
参考文献:略


图 书 信 息

《疫苗创新技术》
主编:王佑春
开本:大16开
规格:210*285mm
页数:932页
定价:290.00元

购书链接
https://dy.chinahlyy.com/web/Page?_pid=d_show_product&product_id=123


PART.01

PART.02
1.微信订阅:扫描二维码进行订阅。

2.PC端网络订阅:登录官网(www.cfdam-health.com)“征订”窗口进行订阅。
3.填表订阅:请完整填写订阅单,连同汇款转账凭证(可另附页)发送邮件至杂志本部联系人,即完成订阅。

扫描二维码即可下载征订单

喜欢就请点个"在看"吧~

标签:药品